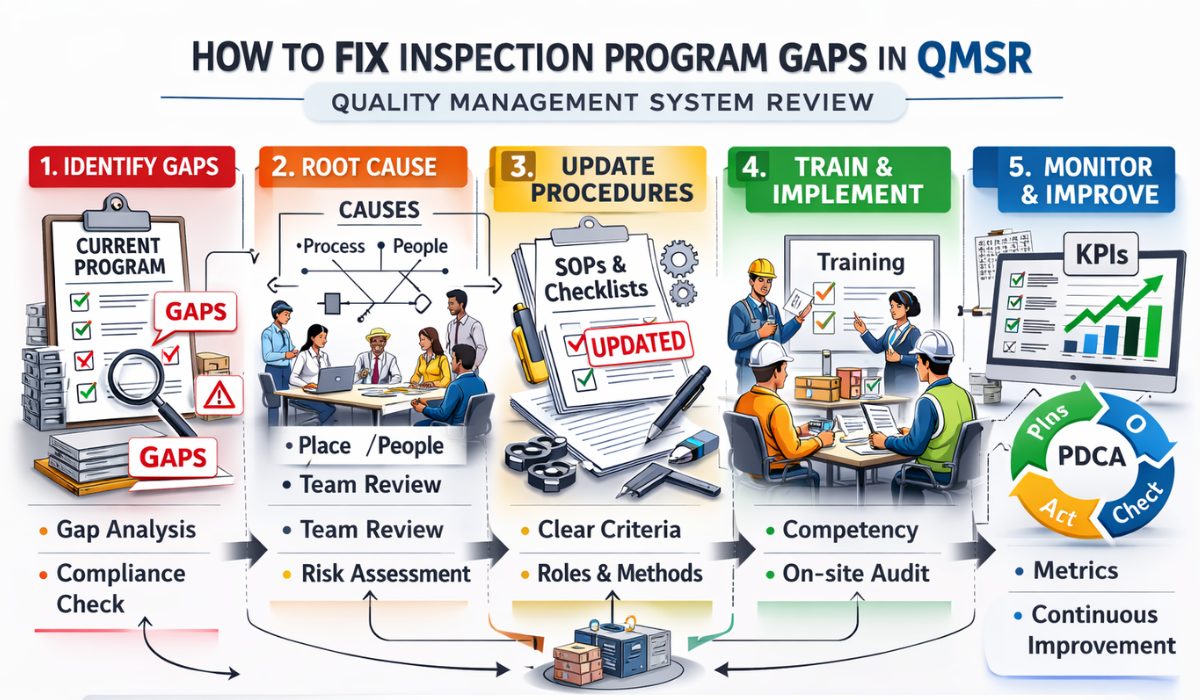
How To Fix Inspection Program Gaps In QMSR
The transition from the Quality System Regulation (QSR) to the Quality Management System Regulation (QMSR) has introduced a paradigm shift in how the FDA evaluates medical device manufacturers. Central to this change is the alignment with ISO 13485:2016, which emphasizes a risk-based approach to quality. However, many organizations still struggle with legacy processes, leading to significant inspection program gaps. Learning how to fix inspection program gaps in QMSR is not just a matter of compliance; it is a strategic necessity to ensure product safety and operational continuity.
An effective inspection program serves as the first line of defense against regulatory non-compliance. When gaps exist in your internal audit or inspection framework, critical failures in design controls or corrective actions can go unnoticed until an official FDA inspection occurs. To bridge these gaps, manufacturers must systematically evaluate their current state against the new QMSR requirements, focusing on documentation, personnel training, and the integration of risk management throughout the product lifecycle.
Strategic Alignment with FDA QMSR
The first step in addressing deficiencies is understanding the FDA QMSR Inspection Program Rule Interpretation. The FDA now places a heavier reliance on the manufacturer’s ability to demonstrate that their quality management system is not only documented but effectively implemented. This means that “paper-based compliance” is no longer sufficient. Fix inspection program gaps by ensuring that your internal inspectors are trained to look for evidence of effectiveness rather than just the presence of a signature.
Identifying Common Program Gaps
Before you can fix inspection program gaps in QMSR, you must identify where they originate. Most gaps in medtech firms occur due to inconsistent data silos or a lack of objective evidence during the inspection process. For instance, many companies fail to link their risk management files directly to their inspection criteria. This misalignment often results in inspectors overlooking high-risk components while over-auditing low-risk administrative processes.
Common QMSR Inspection Program Challenges For Medtech often include inadequate root cause analysis and a failure to monitor the effectiveness of previous corrective actions. If your internal inspection program does not verify that a CAPA (Corrective and Preventive Action) actually solved the problem, the gap remains open. To address this, your inspection checklists must be dynamic, reflecting recent non-conformances and emerging trends in your production data.
Utilizing Standardized Frameworks
To streamline the identification process, many experts recommend using a Medical Device QMSR Inspection Program Template. A template provides a structured baseline, ensuring that no critical area—from management responsibility to resource provision—is ignored. By comparing your existing audit results against a standardized template, you can visually see where your program falls short of the ISO 13485:2016 requirements adopted by the QMSR.
Best Practices for Closing the Gaps
Fixing gaps requires a proactive culture rather than a reactive one. This starts with an Introduction to QMSR Compliance Best Practices. One of the most effective strategies is the implementation of “Vertical Audits,” where an inspector follows a single product from the raw material stage through to final distribution. This method is far more likely to uncover systemic gaps in the inspection program than a traditional horizontal audit that only looks at one department at a time.
Another essential practice is the periodic rotation of internal auditors. When the same individual audits the same department for years, “habitual blindness” sets in. By bringing in a fresh set of eyes—or even an external consultant—you can identify gaps that have become part of the daily routine. Ensure that all findings are documented in a centralized QMS software to maintain data integrity and traceability.
Technical Implementation of Controls
The Best Practices For Medical Device Inspection Program QMSR Compliance suggest that automation is key. Manual inspection logs are prone to errors and are difficult to trend. By digitizing your inspection program, you can use real-time analytics to spot “drifts” in quality before they become significant gaps. Automated alerts can notify management when an inspection is overdue or when a specific gap has been identified across multiple sites.
Top Tools for QMSR Inspection Success
To effectively fix inspection program gaps in QMSR, you need the right technology stack. The market offers various Top inspection Program Tools For QMSR Compliance that facilitate everything from mobile auditing to automated report generation. These tools ensure that your data is captured accurately and is immediately available for review during a formal FDA inspection.
Key features to look for in these tools include:
- Risk-based scheduling modules.
- Offline mobile audit capabilities.
- Direct integration with CAPA and Change Control systems.
- Real-time dashboarding for management review.
Detailed Gap Analysis and Resolution
When you fix inspection program gaps in QMSR, you must look at the “Process Approach.” This approach treats the quality system as a series of interlinked processes. If one process—such as supplier management—has a gap, it will inevitably affect the inspection results of the final product. Your gap resolution plan should include a detailed timeline, assigned responsibilities, and specific metrics for success.
For example, if a gap is found in the “Design Transfer” stage, the fix isn’t just to update a SOP (Standard Operating Procedure). The fix must involve training the production team, updating the inspection jigs, and validating that the new inspection criteria can actually detect the defects identified in the design risk assessment. This holistic view is what the FDA expects under the new QMSR framework.
Training and Competency
A frequently overlooked gap is the competency of the inspectors themselves. Under QMSR, inspectors must demonstrate an understanding of risk management principles. Providing regular training sessions on ISO 14971 (Risk Management for Medical Devices) is crucial. If your inspectors don’t understand the “why” behind an inspection point, they won’t be able to effectively identify a gap when they see one.
Conclusion
To fix inspection program gaps in QMSR is a continuous journey of improvement. As the FDA continues to harmonize its requirements with international standards, medical device manufacturers must remain vigilant. By moving away from checklist-based auditing and toward a risk-based, process-oriented inspection program, organizations can not only ensure compliance but also drive significant improvements in product quality and patient safety. Start with a thorough gap analysis, leverage modern digital tools, and foster a culture of quality that transcends mere regulatory requirements.
FAQs
1. What is the biggest difference between QSR and QMSR for inspections? The biggest difference is the formal incorporation of ISO 13485:2016. While QSR was similar, QMSR explicitly requires a risk-based approach across all quality management processes, making the inspection of risk files a central focus.
2. How often should I perform a gap analysis on my inspection program? It is recommended to perform a formal gap analysis at least annually or whenever there is a significant change in regulatory requirements, product portfolio, or manufacturing processes.
3. Can I use my old QSR checklists for QMSR inspections? While many elements remain the same, old checklists usually lack the specific “Risk Management” and “Resource Management” focus required by QMSR. You should update your checklists to reflect the ISO 13485:2016 structure.
4. What are the consequences of failing to fix inspection program gaps? Unresolved gaps typically lead to Form 483 observations during FDA inspections. If these are not addressed, they can escalate into Warning Letters, consent decrees, or even product recalls.
5. How do I handle gaps found in my supplier’s inspection program? Under QMSR, you are responsible for the quality of your suppliers. You must conduct supplier audits and ensure their inspection programs align with your quality requirements through formal Quality Agreements.
6. Is software validation required for inspection program tools? Yes, under both 21 CFR Part 11 and QMSR, any software used to manage quality processes, including inspection programs, must be validated for its intended use to ensure data integrity.
References
- FDA QMSR Final Rule: https://www.federalregister.gov – The official government document detailing the transition from QSR to QMSR and the adoption of ISO 13485.
- ISO 13485:2016 Standard: https://www.iso.org – The international standard for medical device quality management systems that forms the core of the new QMSR.
- FDA Inspectional Strategy (QSIT): https://www.fda.gov – Guidance on how the FDA conducts quality system inspections and what inspectors look for in a program.
- Global Harmonization Working Party (GHWP): https://www.ghwp.info – Provides resources on international regulatory alignment for medical devices and inspection best practices.
- Medical Device Innovation Consortium (MDIC): https://mdic.org – A public-private partnership that offers tools and whitepapers on improving quality and compliance in the medtech industry.
- RAPS Regulatory Focus: https://www.raps.org – A professional resource providing expert analysis on QMSR implementation challenges and gap remediation strategies.









